
Research Overview
A sample of current research topics in the Computational Materials Research group:
2D Metal-Organic Networks on Functional Surfaces

|

|
When placed on an atomically flat surface, individual atoms and small molecules can spontaneously self-assembly into highly ordered two-dimensional architectures whereby the magnetic atoms are distributed in a ordered network separated by the organic molecules.
Such networks are promising candidates for applications in ultra-high density data storage, chemical sensing and as quantum spin systems for quantum computing.
As they are only few-atoms thick, the interaction between the individual components and the surface is critical to both the self-assembly process and the
resulting network properties.
We are using advanced computational techniques to extract the driving factors influencing the interactions between a surface and the metal-organic network.
We are investigating how functional substrates with switchable electrical and magnetic polarization can be used to control the structure and behaviour of such networks. We will use this information to design optimized networks for data storage and quantum computing.
Such networks are promising candidates for applications in ultra-high density data storage, chemical sensing and as quantum spin systems for quantum computing.
As they are only few-atoms thick, the interaction between the individual components and the surface is critical to both the self-assembly process and the
resulting network properties.
We are using advanced computational techniques to extract the driving factors influencing the interactions between a surface and the metal-organic network.
We are investigating how functional substrates with switchable electrical and magnetic polarization can be used to control the structure and behaviour of such networks. We will use this information to design optimized networks for data storage and quantum computing.
Modelling multi-valent ion intercalation in MXene materials for next-generation energy storage
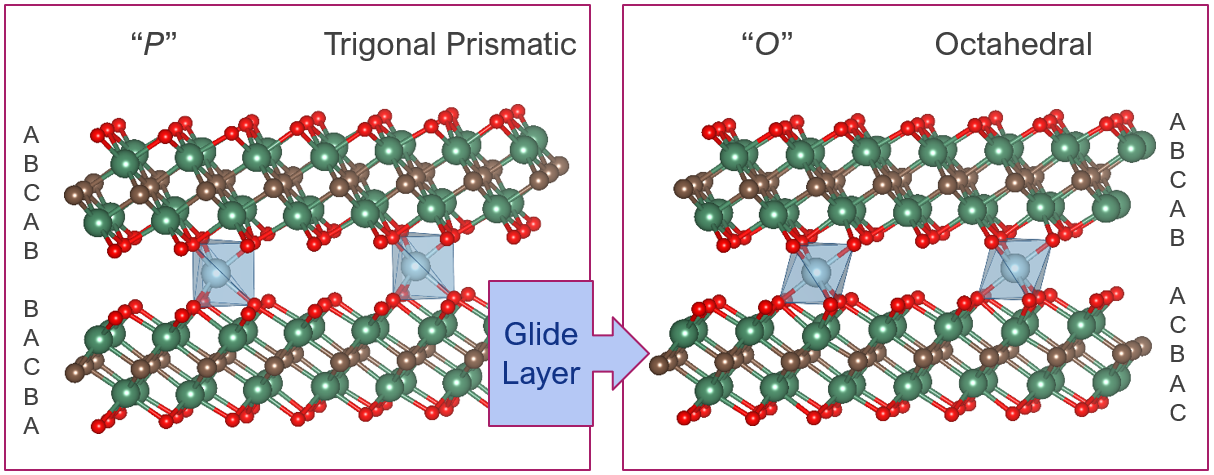
Multi-valent ion batteries, including Mg2+, Ca2+, Zn2+, and Al3+-based batteries, are potentially safer, cheaper, and have higher energy densities than those of commercial Li-ion batteries (LIBs).
However, finding electrode materials capable of reversibly intercalating these cations is a major stumbling block to progress in this area. While graphite is the most popular commercial rechargeable battery anode it is only suitable for Li-ion based batteries as its interlayer spacing is too small to accommodate larger ions.
We are investigating the suitability of a recently discovered family of 2D materials, namely MXene materials, for this purpose.
However, finding electrode materials capable of reversibly intercalating these cations is a major stumbling block to progress in this area. While graphite is the most popular commercial rechargeable battery anode it is only suitable for Li-ion based batteries as its interlayer spacing is too small to accommodate larger ions.
We are investigating the suitability of a recently discovered family of 2D materials, namely MXene materials, for this purpose.
Structural and Orientational Ordering of Solvent Molecules in Graphene Dispersions

|

|
Liquid phase exfoliation (LPE) is the most promising method for the low-cost, scalable production of two-dimensional nanosheets from their bulk counterparts.
Extensive exfoliation occurs in most solvents due to the huge amount of energy introduced by sonication or shear mixing. However, the subsequent dispersion is not always stable, with extensive reaggregation occurring in some solvents.
Identifying the optimal solvent for a particular layered material requires a fundamental understanding of the mechanism involved in maintaining a stable dispersion.
We are using molecular dynamics and ab initio calculations to systematically study the origin of the solvent dependence of the stabilization of exfoliated monolayers.
Extensive exfoliation occurs in most solvents due to the huge amount of energy introduced by sonication or shear mixing. However, the subsequent dispersion is not always stable, with extensive reaggregation occurring in some solvents.
Identifying the optimal solvent for a particular layered material requires a fundamental understanding of the mechanism involved in maintaining a stable dispersion.
We are using molecular dynamics and ab initio calculations to systematically study the origin of the solvent dependence of the stabilization of exfoliated monolayers.
One-dimensional Moiré physics in heterostrained bilayer graphene

Modulated stacking profiles in bilayer graphene systems lead to a wide range of new properties, particularly when combined with an interlayer bias.
Such a bias will open a band gap in regions with particular stackings, leading to a complex distribution of gapped and conducting regions which follow the underlying stacking pattern.
This has been widely studied in twisted bilayers where, for example, networks of 1D topological channels have been found. The localisation of electronic
states in different regions of the lattice, together with the resulting strong interaction effects, are connected with the formation of correlated insulating states and unconventional superconductivity in twisted systems.
Heterostrained, untwisted BLG could provide a viable alternative to twistronics to engineer topological and exotic physical phenomena in such systems. We are using ab initio and tight-binding calculations to determine whether heterostrain can be used to reversibly engineer the stacking order in a graphene bilayer and to investigate the emergent Moiré features.
Such a bias will open a band gap in regions with particular stackings, leading to a complex distribution of gapped and conducting regions which follow the underlying stacking pattern.
This has been widely studied in twisted bilayers where, for example, networks of 1D topological channels have been found. The localisation of electronic
states in different regions of the lattice, together with the resulting strong interaction effects, are connected with the formation of correlated insulating states and unconventional superconductivity in twisted systems.
Heterostrained, untwisted BLG could provide a viable alternative to twistronics to engineer topological and exotic physical phenomena in such systems. We are using ab initio and tight-binding calculations to determine whether heterostrain can be used to reversibly engineer the stacking order in a graphene bilayer and to investigate the emergent Moiré features.
